Currently, genome-wide association studies (GWAS) are an area of focus for many PGRN sites. Over the course of the next five years, PGRN sites will likely generate data via exomic or whole-genome sequencing as well as epigenomics and high-throughput data generation platforms. Methods developed by this resource will stay current with the needs of the PGRN. We currently have three proposed areas of methodological development; recognizing that these are intended to be examples of the methods developed in the context of pharmacogenomic studies, and that novel methods for analysis of pharmacogenomic phenotypes will continue over the 5-year cycle.

The PAAR group at the University of Chicago has developed a database and web site to facilitate annotation of SNPs and CNVs (http://scandb.org) that provides not only physical, functional, linkage disequilibrium (LD) annotations, but also serves results of qQTL studies that we have conducted in the HapMap CEU and YRI lymphoblastoid cell lines (LCLs)2-5 as an adjunct to PAAR studies on the cytotoxicity of chemotherapeutic agents2-5. Thus, using the SCAN database, it is possible to enter a list of SNPs - the top signals from a genome-wide association study (GWAS), for example - and obtain physical, functional, LD annotations for each SNP, as well as an indication of distant and local transcript levels for which that SNP is a significant predictor.

The group at St. Jude Children's Research Hospital will develop a methodology to integrate various types of genomic data (SNP genotypes, CNVs, mRNA expressions, etc.) and multiple related phenotypes into coherent genomic association networks (CGANs) that reveal inter-relationships among different types of genomic features and their associations with (effects on) phenotypes of interest in pharmacogenomics studies. The molecular mechanism underlying inter-related, multiple phenotypes can be a combination of effects from different genomic components at different levels. Thus integrated analyses combining various genomic features can help elucidate further the underlying biology than single-feature screening methods.
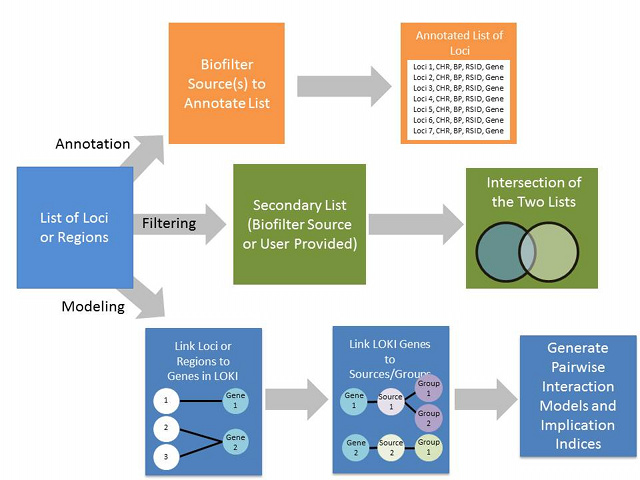
Biofilter is a tool for knowledge-driven multi-SNP analysis of large scale SNP data using information from public databases (The Gene Ontology, The Database of Interacting Proteins, The Protein Families Database, The Kyoto Encyclopedia of Genes and Genomes, Reactome, and Biopath). As part of P-STAR, we will continue to develop the Biofilter resource. In its current implementation, it was primarily developed for common disease GWAS data. There are a number of knowledge sources that would primarily be important to include for pharmacogenomics studies in particular including: PharmGKB VIP genes/pathways, DMET pathways, and eQTL data. We will integrate these and other relevant additional sources of information into the Biofilter.
A network resource for coordination of statistical analysis and methods development in the PGRN.